Phénylcétonurie
Marian Grosser a étudié la médecine humaine à Munich. De plus, le docteur, qui s'intéressait à beaucoup de choses, osa faire des détours passionnants : étudier la philosophie et l'histoire de l'art, travailler à la radio et, enfin, aussi pour un Netdoctor.
En savoir plus sur les experts Tout le contenu de est vérifié par des journalistes médicaux.La phénylcétonurie (PCU) est une maladie congénitale et héréditaire du métabolisme des protéines. Il empêche la dégradation de l'acide aminé phénylalanine. Cela s'accumule dans le corps et perturbe le développement du cerveau de l'enfant. Si elle n'est pas traitée, la phénylcétonurie entraîne de graves déficiences intellectuelles. Avec un traitement opportun, cependant, les patients peuvent mener une vie normale. Tout savoir sur la phénylcétonurie ici !
Codes CIM pour cette maladie : Les codes CIM sont des codes internationalement reconnus pour les diagnostics médicaux. On les retrouve, par exemple, dans les lettres des médecins ou sur les certificats d'incapacité de travail. E70
Phénylcétonurie: description
La phénylcétonurie (PCU) est une maladie métabolique héréditaire qui existe dès la naissance et interfère avec la dégradation de l'acide aminé essentiel phénylalanine. Les acides aminés sont les éléments constitutifs de base des protéines et donc des composants métaboliques vitaux. Certains d'entre eux ne peuvent pénétrer dans le corps que par la nourriture, l'organisme ne peut pas les produire lui-même. Ces acides aminés sont appelés essentiels.
Que se passe-t-il avec la phénylcétonurie?
Normalement, les acides aminés sont soumis à un équilibre entre absorption / accumulation et dégradation, de sorte qu'il y en ait toujours autant que le corps a besoin. Dans le cas de divers acides aminés, une carence ou un excès peut causer des dommages considérables et provoquer divers symptômes.
Dans la PCU dite classique, l'effet de la phénylalanine hydroxylase (PAH) est limité voire totalement absent. En raison de la carence en PAH, la phénylalanine s'accumule de plus en plus dans le corps. Une concentration trop élevée de phénylalanine perturbe considérablement le développement du cerveau et conduit à des déficiences intellectuelles chez les jeunes patients à un stade précoce.
Étant donné que la dégradation normale de la phénylalanine n'est pas possible avec la maladie, d'autres produits de dégradation, appelés phénylcétones, se forment. Ils sont excrétés dans l'urine et sont responsables du nom de la maladie.
Phénylcétonurie atypique
Même avec des formes atypiques de phénylcétonurie, la dégradation de la phénylalanine est perturbée. Cependant, la cause n'est pas un défaut de l'HTAP. Au lieu de cela, la fonction d'une coenzyme, la tétrahydrobioptérine (BH4), est restreinte. Il est indirectement impliqué dans la dégradation de la phénylalanine car le PAH a besoin de BH4 pour convertir la phénylalanine en tyrosine.
Étant donné que la BH4 est également importante pour la production des substances messagères dopamine et sérotonine, la phénylcétonurie atypique avec déficit en BH4 est généralement plus compliquée que la forme classique.
Qui la phénylcétonurie affecte-t-elle?
La PCU est l'une des maladies métaboliques congénitales les plus courantes. On estime qu'environ un nouveau-né sur 7 000 dans le monde le développera, sans différence entre les filles et les garçons. Comme il s'agit d'une maladie héréditaire, plusieurs membres d'une même famille sont souvent touchés.
Phénycétonurie: symptômes
Au début, les bébés atteints de phénylcétonurie ne présentent aucun symptôme de la maladie. Les premiers problèmes ne surviennent qu'entre le quatrième et le sixième mois de vie, si la maladie n'a pas encore été reconnue et traitée. Surtout, la maturation cérébrale perturbée entraîne des complications massives au fil du temps. Les symptômes de la PCU non traitée comprennent :
- un fort déficit mental. Les lésions cérébrales progressent jusqu'à la puberté puis stagnent. Les enfants atteints sont alors généralement gravement handicapés mentaux.
- Crises (convulsions) (épilepsie). En raison des dommages, les cellules nerveuses du cerveau sont particulièrement sensibles et surexcitables. Il en résulte de fréquentes crises d'épilepsie.
- handicaps moteurs. Non seulement les cellules du cerveau, mais aussi les muscles du patient peuvent être surexcités. C'est pourquoi il devient souvent tendu (spasticité), ce qui entraîne divers troubles du mouvement.
- Troubles du comportement. Certains enfants atteints de phénylcétonurie sont hyperactifs et inhabituellement agressifs, et les accès de colère sont également plus fréquents.
- une petite tête (microcéphalie). Parce que le cerveau du patient ne se développe pas correctement, la croissance de la tête prend également du retard. La petite circonférence de la tête par rapport à leurs pairs est particulièrement visible chez les enfants plus âgés.
- une odeur perceptible. La PKU produit certains produits de dégradation de la phénylalanine qui sentent comme les excréments de souris. Ces substances sont principalement excrétées dans les urines, mais aussi en partie à travers la peau.
- changements cutanés semblables à l'eczéma
Parce que la production du pigment mélanine est également perturbée dans la phénylcétonurie, de nombreuses personnes atteintes ont une peau très claire et sensible au soleil et des cheveux blonds blancs. L'iris des yeux est également bleu clair à transparent et laisse transparaître le fond rougeâtre.
Les symptômes de la PCU varient en gravité d'une personne à l'autre. La principale raison en est que l'activité de la phénylalanine hydroxylase (PAH) est limitée différemment chez chaque patient. Certains ont encore une certaine activité résiduelle, de sorte que moins de phénylalanine s'accumule dans l'organisme. D'autres ne montrent aucune activité enzymatique - la maladie progresse d'autant plus rapidement et plus sérieusement.
Phénylcétonurie : causes et facteurs de risque
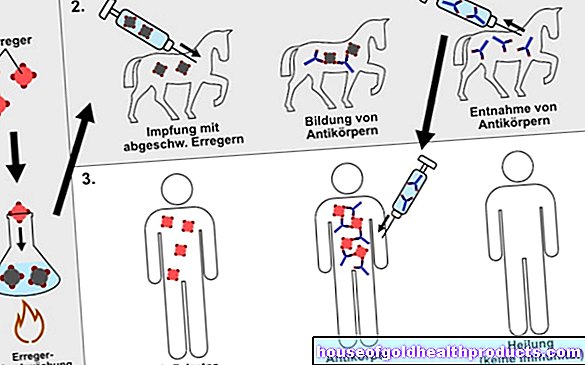
La phénylcétonurie est une maladie héréditaire. De nombreuses mutations génétiques sont maintenant connues qui conduisent à un défaut de l'HTAP. Le type de mutation détermine dans quelle mesure la dégradation de la phénylalanine est limitée.
La PCU est héritée de manière récessive, ce qui signifie qu'une personne peut être porteuse d'un gène altéré sans développer la maladie. De même, les personnes atteintes de phénylcétonurie peuvent engendrer des enfants en bonne santé.
Ce n'est que si les deux parents ont des mutations dans leur constitution génétique qu'il y a une certaine probabilité que leur progéniture développe une phénylcétonurie. Si les parents sont tous les deux non seulement porteurs des gènes, mais qu'ils sont également atteints de PCU, tous les enfants ensemble la développeront également.
Phénylcétonurie : examens et diagnostic
Étant donné que les conséquences graves de la phénylcétonurie peuvent être évitées en commençant le traitement à temps, il est particulièrement important de découvrir la maladie le plus tôt possible. En Allemagne, les enfants sont examinés pour diverses maladies congénitales, y compris la PCU, dans le cadre d'un examen général (dépistage néonatal) le troisième jour après la naissance.
Spectrométrie de masse en tandem
De nombreux troubles métaboliques congénitaux sont désormais diagnostiqués à l'aide de la spectrométrie de masse en tandem. Il permet d'examiner rapidement et facilement le sang du nouveau-né. En plus de la phénylcétonurie, les médecins peuvent détecter plus de 20 autres maladies en quelques minutes.
Test de Guthrie
Le test de Guthrie, du nom de son inventeur, permet également de diagnostiquer la PCU. Pour ce faire, une petite quantité de sang est prélevée sur le talon de l'enfant et appliquée sur un morceau de papier filtre. En laboratoire, vous pouvez alors déterminer si la concentration de phénylalanine est augmentée.
Le test de Guthrie a été introduit dans les années 1960 et a longtemps été la méthode standard pour diagnostiquer la phénylcétonurie. Cependant, elle présente des inconvénients par rapport à la spectrométrie de masse en tandem. Le test de Guthrie ne donne un résultat qu'après cinq jours, ce qui est également sujet aux erreurs. Par exemple, des facteurs tels que l'alimentation de l'enfant ou une éventuelle antibiothérapie faussent les résultats. Dans certains pays, le test de Guthrie est encore utilisé, en Allemagne, il n'est généralement plus utilisé.
D'autres épreuves
Si le dépistage néonatal révèle une suspicion de phénylcétonurie, un autre examen suit pour confirmation. Cela peut également être utilisé pour déterminer la concentration exacte de phénylalanine dans le sang.
Enfin, il est important de distinguer s'il s'agit d'une PCU typique (classique) ou atypique. Des tests spéciaux sont également disponibles pour cela, comme le test de stress à la tétrahydrobioptérine. La distinction est importante car la phénylcétonurie atypique est traitée différemment de la forme classique.
Examen du liquide amniotique
La phénylcétonurie peut déjà être diagnostiquée pendant la grossesse (diagnostic prénatal). Pour ce faire, une petite quantité de liquide amniotique est prélevée dans le sac amniotique de la mère et les cellules de l'enfant à naître qu'il contient sont examinées. Tous les défauts génétiques qui causent la PCU peuvent être identifiés de cette manière.
Avec le dépistage néonatal, cependant, la phénylcétonurie est généralement découverte suffisamment tôt pour être traitée. Étant donné qu'un test de liquide amniotique est toujours associé à un certain risque, son utilisation pour le diagnostic de la PCU n'est généralement pas utile.
Phénylcétonurie : traitement
Il n'y a qu'une seule façon de contrer l'excès de phénylalanine dans la PCU : les enfants atteints doivent suivre un régime alimentaire spécial afin d'absorber le moins possible de phénylalanine avec leur alimentation. Ils ont également besoin de certains compléments alimentaires pour remplacer les substances qui seraient formées à partir de la phénylalanine chez les enfants en bonne santé.
Le traitement doit commencer avant l'apparition des premiers symptômes d'un trouble du développement, c'est-à-dire dans les deux premiers mois de la vie. Les lésions cérébrales qui se sont déjà produites ne peuvent pas être inversées.
Thérapie nutritionnelle avec le régime PCU
Toutes les protéines naturelles sont constituées d'environ cinq pour cent de l'acide aminé phénylalanine. La plupart des aliments en contiennent bien plus que ce dont le corps a besoin. Ce n'est pas un problème pour une personne en bonne santé car elle se décompose et excrète les quantités excessives de phénylalanine.
Les patients atteints de phénylcétonurie, en revanche, doivent se passer pour la plupart de protéines naturelles. Des produits spéciaux fabriqués industriellement remplacent les composants alimentaires manquants. D'une part, on veut éviter un excès de phénylalanine, d'autre part, il faut approvisionner le patient en substances qui feraient autrement défaut, comme la tyrosine, qui est formée à partir de la phénylalanine.
Cependant, l'objectif du régime PCU n'est pas d'arrêter complètement l'absorption de la phénylalanine. Parce que l'organisme a besoin d'une certaine quantité d'acides aminés pour des processus métaboliques importants. Atteindre la bonne concentration représente un grand défi pour les médecins et les patients et nécessite beaucoup de discipline.
Le régime doit donc être respecté toute la vie, mais surtout strictement jusqu'à l'âge de six ans. Car le cerveau se développe très fortement jusqu'à cet âge et est donc particulièrement sensible aux dommages. À l'âge adulte, des concentrations élevées de phénylalanine ne causent pas de lésions cérébrales comme elles le font chez les enfants. Cependant, ils peuvent être le déclencheur d'autres troubles neurologiques tels qu'une mauvaise concentration ou des réactions ralenties.
Le traitement diététique de la phénylcétonurie doit commencer dans un centre spécialisé dans les maladies métaboliques. Parce qu'il n'est pas possible dans toutes les cliniques d'instruire les parents sur le régime alimentaire. Ceci est mieux fait avec l'aide de conseils diététiques, qui montrent comment suivre le régime et surveiller régulièrement les niveaux de phénylalanine dans le sang.
Traitement de la phénylcétonurie atypique
Dans les formes atypiques de PCU, la coenzyme BH4 manquante et certaines substances messagères telles que la dopamine et la sérotonine sont artificiellement remplacées. Dans certains cas, le patient doit également suivre un régime pauvre en phénylalanine.
Phénylcétonurie pendant la grossesse
Si vous êtes enceinte et que vous souffrez vous-même de phénylcétonurie, vous devez tenir compte des éléments suivants :
- Tenez-vous en à votre régime alimentaire de manière particulièrement stricte !
- Faites vérifier vos valeurs sanguines de phénylalanine à intervalles fréquents. Parce que les concentrations de phénylalanine chez l'enfant à naître sont environ deux fois plus élevées que chez la femme enceinte. En plus du cerveau, ils peuvent également endommager le cœur et les yeux de l'enfant à naître. Des malformations du squelette de l'enfant peuvent également résulter de taux élevés de phénylalanine pendant la grossesse.
- Afin d'exclure les lésions cérébrales chez l'enfant au début de la grossesse, les femmes atteintes de phénylcétonurie doivent planifier soigneusement leur grossesse et prendre des précautions dès le début.
- Chez les femmes enceintes atteintes de phénylcétonurie, un régime alimentaire non strictement respecté peut entraîner une fausse couche (avortement spontané).
- Dans tous les cas, les femmes enceintes atteintes de phénylcétonurie doivent demander conseil à un médecin.
Phénylcétonurie: évolution de la maladie et pronostic
Si la phénylcétonurie est diagnostiquée précocement, si possible chez le nouveau-né, et si le régime spécial PCU est suivi, le pronostic est généralement bon. Les enfants se développent spirituellement et ont une espérance de vie moyenne.
Cependant, si elles ne sont pas traitées, les lésions cérébrales entraînent de graves troubles du développement mental qui ne peuvent être corrigés plus tard. Les personnes touchées ont une espérance de vie normale, mais leur quotient intellectuel est presque toujours très inférieur à la norme.
La forme atypique rare de phénylcétonurie, dans laquelle un déficit en BH4 est présent, est un cas particulier. Cette variante de la phénylcétonurie peut entraîner des lésions neurologiques progressives avec des crampes sévères malgré le régime.
Mots Clés: valeurs de laboratoire pieds sains grossesse naissance










.jpg)