Syndrome d'Angelman
Lisa Vogel a étudié le journalisme départemental avec une spécialisation en médecine et en biosciences à l'Université d'Ansbach et a approfondi ses connaissances journalistiques dans le cadre d'un master en information et communication multimédia. Cela a été suivi d'un stage dans l'équipe éditoriale de Depuis septembre 2020, elle écrit en tant que journaliste indépendante pour
Plus de messages par Lisa Vogel Tout le contenu de est vérifié par des journalistes médicaux.Le syndrome d'Angelman (syndrome de la marionnette heureuse) est une maladie génétique rare. Elle se manifeste, entre autres, par des limitations mentales et physiques, des troubles du développement (notamment du langage) et de l'hyperactivité. Ce qui frappe, c'est l'apparence de poupée et l'expression faciale heureuse des personnes touchées. En savoir plus sur la maladie rare !
Codes CIM pour cette maladie : Les codes CIM sont des codes internationalement reconnus pour les diagnostics médicaux. On les retrouve, par exemple, dans les lettres des médecins ou sur les certificats d'incapacité de travail. Q93

Bref aperçu
- Qu'est-ce que le syndrome d'Angelman ? Maladie génétique rare qui se manifeste par des limitations mentales et physiques dans le développement de l'enfant
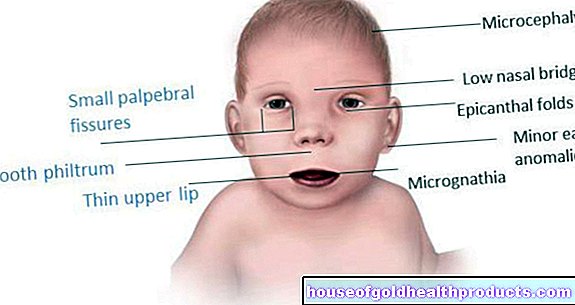
- Symptômes : traits du visage ressemblant à des poupées, trouble du développement, troubles de la coordination, pas ou presque pas de développement du langage, intelligence réduite, crises d'épilepsie, rires sans fondement, crises de rire, bave excessive, agitation joyeuse des bras
- Causes : défaut génétique sur le chromosome 15
- Diagnostic : entre autres conversation, examen physique, examens génétiques
- Thérapie : Aucune thérapie causale disponible ; Soutien, par exemple physiothérapie, orthophonie, ergothérapie ; éventuellement des médicaments pour soulager les symptômes (par exemple en cas de convulsions)
- Prévision : espérance de vie normale ; pas de vie indépendante possible
Syndrome d'Angelman : définition
Le syndrome d'Angelman (SA) est causé par une anomalie génétique sur le chromosome 15. Cette anomalie perturbe le développement physique et mental des personnes atteintes. Les troubles du développement de la parole, l'insécurité motrice et un visage heureux sont les symptômes les plus visibles du syndrome d'Angelman.
Le nom "Syndrome d'Angelman" vient du découvreur de la maladie, le pédiatre anglais Harry Angelman. En 1965, il a comparé les images cliniques de trois enfants qui avaient des traits de visage de poupée. Les enfants ont beaucoup ri et ont fait des mouvements saccadés - comme des marionnettes. Cela a conduit au nom anglais « Happy Puppet Syndrome »(happy puppet).
Le syndrome d'Angelman survient chez les deux sexes. Le risque de la maladie est d'environ 1: 20 000. Cela fait du syndrome une maladie rare.
Syndrome d'Angelman : Symptômes
À la naissance, les enfants atteints du syndrome d'Angelman sont normaux. Les troubles du développement moteur et cognitif ne deviennent de plus en plus perceptibles dans la petite enfance et la petite enfance. Les caractéristiques de la maladie génétique sont :
- développement moteur retardé
- coordination altérée
- souvent pas ou presque pas de développement du langage
- diminution de l'intelligence
- comportement hyperactif et fougueux
- rire sans fondement
- Crises de rire
- gestes joyeux (par exemple, agiter les bras)
- bave excessive
- tir fréquent de la langue
Certains enfants atteints du syndrome d'Angelman ont également :
- Microcéphalie (tête anormalement petite) - pas à la naissance, mais au fur et à mesure qu'elle se développe
- Saisies
- Modifications de l'activité électrique du cerveau
- peau et yeux très clairs en raison d'une diminution de la pigmentation (hypopigmentation)
- Louche (strabisme)
Syndrome d'Angelman: causes
La cause du syndrome d'Angelman est une anomalie génétique sur le chromosome 15 : la fonction du gène UBE3A est altérée chez les personnes atteintes. Ce gène produit normalement une enzyme qui est impliquée dans la décomposition des protéines endommagées ou superflues dans les cellules. Il aide ainsi la cellule à fonctionner normalement.
Le gène UBE3A est situé dans la région chromosomique 15q11q13. Là, les gènes sont soumis à ce que l'on appelle « l'empreinte génomique ». Cela signifie qu'ils ne sont actifs que sur l'un des chromosomes parentaux (dans nos cellules corporelles, il y a deux copies de chaque chromosome - une de la mère et une du père). Ceci est régulé par un processus chimique - la méthylation : des groupes méthyle attachés à certains points peuvent activer ou désactiver un gène.
Le gène est actif sur les deux chromosomes dans de nombreuses cellules du corps, mais pas dans les cellules nerveuses du cerveau : chez de nombreuses personnes, le gène UBE3A sur le chromosome paternel 15 est désactivé par empreinte. En conséquence, UBE3A n'est actif que sur le chromosome maternel 15 dans le cerveau. Cela signifie également : si la copie du gène maternel présente une erreur, celle-ci ne peut pas être compensée par la copie du gène paternel désaffectée. Et c'est précisément cette combinaison qui se produit dans le syndrome d'Angelman : le segment du gène paternel est éteint, le segment du gène maternel est défectueux.
L'erreur génétique sous-jacente peut être de différents types :
- Suppression : Environ 75 % de toutes les personnes atteintes du syndrome d'Angelman n'ont pas la région pertinente 15q11q13 avec le gène UBE3A sur le chromosome maternel 15. Étant donné que la région correspondante sur le chromosome paternel 15 est « coupée » par l'empreinte, le corps peut utiliser l'enzyme dont le plan de construction est enregistré dans Gen UBE3A, ne créez pas.
- Mutation du gène UBE3A : Une modification spontanée du gène entraîne la perte des informations qu'il contient. Cela est vrai de cinq à dix pour cent des personnes atteintes du syndrome d'Angelman. Dans environ un cas sur cinq, il y a une mutation familiale : ici, la mère porte déjà le changement génétique sur le chromosome de son père.
- Deux chromosomes paternels 15 : La personne affectée a hérité des deux chromosomes 15 de son père, aucun de la mère (médicalement appelée « disomie uniparentale paternelle 15 »). Il n'y a donc pas de gène UBE3A actif. Cela s'applique à environ un à deux pour cent de tous les patients atteints du syndrome d'Angelman.
- Erreur d'empreinte : Le gène UBE3A sur le chromosome maternel 15 - comme celui sur le chromosome paternel 15 - est désactivé par empreinte. De plus, une certaine partie du chromosome peut être manquante (suppression). Une erreur d'impression se trouve dans un à quatre pour cent des cas de syndrome d'Angelman.
Dans les 10 à 15 % des cas restants, la cause du syndrome d'Angelman est inconnue. Soit dit en passant : si le gène maternel est désactivé et que le gène paternel est défectueux, les enfants souffrent du syndrome de Prader-Willi.
Le syndrome d'Angelman est-il héréditaire ?
En général, le risque de récidive du syndrome d'Angelman est faible. Cela signifie le risque que les parents d'un enfant affecté aient d'autres enfants qui ont également le syndrome. Dans certains cas, cependant, ce risque dépend en grande partie du défaut génétique sous-jacent au syndrome d'Angelman.
Par exemple, dans le syndrome d'Angelman dû à deux chromosomes paternels 15 (disomie paternelle uniparentale 15), il est inférieur à un pour cent. D'autre part, le syndrome d'Angelman dû à une erreur d'empreinte avec perte d'un certain segment de gène (délétion IC) peut survenir dans la moitié des cas chez un frère ou une sœur.
Ce risque accru existe également avec une mutation UBE3A - à condition que la mère soit déjà porteuse du défaut génétique (dans environ 20 pour cent de tous les cas de mutation). Dans de tels cas, la mère a hérité de la mutation de son père. UBE3A est donc modifié dans le chromosome paternel de la mère. Si elle est désactivée, la mère ne remarquera pas la mutation. Cependant, elle peut transmettre le chromosome à ses enfants, où il - alors en tant que chromosome maternel - peut provoquer le syndrome d'Angelman.
Théoriquement, les patients atteints du syndrome d'Angelman peuvent se reproduire. Selon le moment où les modifications chromosomiques causales se sont produites (par exemple, déjà pendant le développement des cellules germinales ou immédiatement après la fécondation), le risque est parfois très élevé (jusqu'à 100 %) que les personnes touchées transmettent la maladie. Cependant, il y a un manque de données fiables à ce sujet. Il y a eu un cas isolé en septembre 1999 : la mère, atteinte du syndrome d'Angelman, a transmis la maladie ici.
Syndrome d'Angelman : diagnostic
Si vous remarquez les symptômes décrits ci-dessus chez votre enfant, le pédiatre est le premier point de contact. Il pourra cerner plus précisément les causes possibles et vous orienter, vous et votre enfant, vers un spécialiste si nécessaire.
anamnèse
La première étape du diagnostic est un historique médical complet. Le médecin vous posera diverses questions concernant votre enfant, telles que :
- Quels changements avez-vous remarqué chez votre enfant ?
- Votre enfant a-t-il des problèmes physiques ?
- Votre enfant peut-il s'asseoir ?
- Votre enfant attrape-t-il des objets ?
- Votre enfant parle-t-il ?
- Votre enfant est-il souvent visiblement gai ou excité ?
- Votre enfant rit-il dans des situations inappropriées, par exemple lorsqu'il a mal ?
Examen physique
Ceci est suivi de l'examen physique. Le pédiatre teste dans quelle mesure l'enfant développe régulièrement ses capacités motrices et mentales. Des exercices simples sont utilisés à cette fin : par exemple, l'enfant doit se concentrer sur des jouets ou atteindre sélectivement un bloc de construction. Le médecin fait également attention aux expressions faciales de l'enfant. Des rires fréquents, des traits de poupée et la bave sont tous des signes du syndrome d'Angelman.
Si, après l'examen physique, il existe une suspicion de maladie rare, le médecin vous orientera vers un neurologue et un généticien humain.
Tests génétiques
Les tests génétiques sont une partie importante du diagnostic du syndrome d'Angelman. Le médecin a besoin d'un petit échantillon de cellules de l'enfant, qu'il peut obtenir à partir de la muqueuse buccale, par exemple en prélevant un échantillon de sang ou en prélevant un écouvillon. Le matériel génétique (ADN) de ces cellules ou la région chromosomique pertinente 15q11q13 est ensuite examiné plus en détail en laboratoire.
Dans un premier temps, les médecins prêtent attention au schéma de méthylation du segment chromosomique (analyse/test de méthylation). D'autres tests sur les mêmes échantillons (analyse de délétion, analyse de mutation) aident à déterminer plus précisément la cause du syndrome d'Angelman. Pour cela, il peut également être nécessaire d'examiner la constitution génétique des parents. De cette façon, les médecins peuvent déterminer s'il y a déjà un défaut génétique là-bas.
Enquêtes supplémentaires
Des examens complémentaires sont souvent utiles. Par exemple, l'EEG peut être utilisé pour détecter des changements dans l'activité électrique du cerveau, comme cela se produit souvent dans le syndrome d'Angelman. Des examens ophtalmologiques peuvent également être indiqués.
Syndrome d'Angelman : thérapie
Le syndrome d'Angelman est incurable - la cause génétique de la maladie est incurable. Cependant, une intervention précoce peut avoir un effet positif sur le développement moteur et mental des personnes concernées. Une physiothérapie régulière, par exemple, est utile. Il peut améliorer la motricité des enfants, atténuer la mobilité réduite et aider à prévenir les maladies secondaires telles que les déformations de la colonne vertébrale. Les enfants atteints du syndrome d'Angelman bénéficient également d'autres méthodes thérapeutiques telles que l'ergothérapie et l'orthophonie.
De plus, certains symptômes et conditions associés au syndrome d'Angelman peuvent nécessiter un traitement ciblé. Par exemple, les médicaments anticonvulsivants (médicaments antiépileptiques) aident à lutter contre les convulsions et les tranquillisants (sédatifs) aident à soulager les troubles graves du sommeil.
Sur le site Web de Verein Angelman e.V., vous trouverez de nombreuses informations sur le syndrome d'Angelman, des rapports d'expérience et des événements pour les personnes touchées ainsi que des personnes de contact pour les personnes touchées dans toutes les régions d'Allemagne.
Syndrome d'Angelman: évolution de la maladie et pronostic
Première année de vie
Les bébés atteints du syndrome d'Angelman sont plus susceptibles d'avoir des problèmes d'allaitement, de succion et de déglutition. Ils tirent souvent la langue ou bavent beaucoup. De plus, les enfants atteints du syndrome d'Angelman crachent souvent (ce qui est souvent confondu avec une intolérance alimentaire ou une maladie de reflux). Des vomissements fréquents peuvent entraîner une perte de poids dangereuse.
À l'âge de trois à six mois, les enfants atteints du syndrome d'Angelman commencent souvent à sourire. Ils rigolent et gargouillent beaucoup et tirent souvent la langue lors de ces explosions de joie.
Le retard du développement moteur est généralement perceptible entre 6 et 12 mois : les enfants ne rampent pas et ne s'assoient pas. Les mouvements du haut du corps sont souvent instables. Cela rend à son tour la position assise plus difficile.
Une fraction des personnes touchées ont déjà des crises à l'âge de 12 mois.
Un à trois ans
Au cours des trois premières années de la vie, le trouble du développement dans le syndrome d'Angelman devient très évident. Les enfants souffrent davantage de crises bénignes. Certains d'entre eux sont hyperactifs, surexcités et toujours en mouvement. Beaucoup ont tendance à mettre leurs mains ou leurs jouets dans leur bouche tout le temps, ou à tirer fréquemment la langue et à baver. Si les enfants sont particulièrement excités, ils riront souvent excessivement et agiteront les bras tendus.
Le développement du langage avec facultés affaiblies devient apparent pour la première fois à cet âge. Les enfants « babillent » pour eux-mêmes ou crient et couinent, cependant, ne peuvent prononcer que peu ou pas de mots facilement compréhensibles et les utilisent généralement sans contexte.Mais ils comprennent le langage et il y a aussi une interaction sociale avec d'autres personnes.
Puberté et âge adulte
La puberté est souvent en retard de trois à cinq ans chez les enfants atteints du syndrome d'Angelman. Cependant, la maturité sexuelle se développe alors normalement. Il n'y a toujours pas de développement du langage si la compréhension du langage est souvent présente. Les crises à l'âge adulte peuvent généralement être bien gérées avec des médicaments.
Les personnes atteintes du syndrome d'Angelman ont une espérance de vie normale. Une vie indépendante n'est pas possible en raison des limitations mentales.
Mots Clés: systèmes d'organes médecine douce la prévention