hémophilie
et Martina Feichter, rédactrice médicale et biologisteDr. méd. Fabian Sinowatz est pigiste au sein de l'équipe éditoriale médicale de
En savoir plus sur les expertsMartina Feichter a étudié la biologie avec une spécialité pharmacie à Innsbruck et s'est également immergée dans le monde des plantes médicinales. De là, il n'était pas loin d'autres sujets médicaux qui la captivent encore à ce jour. Elle a suivi une formation de journaliste à l'Académie Axel Springer de Hambourg et travaille pour depuis 2007 - d'abord en tant que rédactrice et depuis 2012 en tant que rédactrice indépendante.
En savoir plus sur les experts Tout le contenu de est vérifié par des journalistes médicaux.
L'hémophilie (maladie du sang) est un trouble de la coagulation sanguine qui est généralement héréditaire. Les malades manquent d'importants facteurs de coagulation sanguine ou sont défectueux. En conséquence, les hémophiles sont sujets aux saignements et aux ecchymoses facilement. L'hémophilie n'est pas encore guérie. Grâce aux thérapies modernes, cependant, les hémophiles peuvent mener une vie en grande partie normale. Lisez tout ce que vous devez savoir sur l'hémophilie ici.
Codes CIM pour cette maladie : Les codes CIM sont des codes internationalement reconnus pour les diagnostics médicaux. On les retrouve, par exemple, dans les lettres des médecins ou sur les certificats d'incapacité de travail. D66D67D68
Bref aperçu
- Qu'est-ce que l'hémophilie? Un trouble génétique de la coagulation sanguine (coagulopathie). On l'appelle aussi hémophilie.
- Formes d'hémophilie : La plus courante est l'hémophilie A, suivie de l'hémophilie B. D'autres formes telles que l'hémophilie C, le syndrome de von Willebrand-Jürgens et la parahémophilie sont moins fréquentes.
- Cause : Déficit ou défaut des facteurs de coagulation (protéines sanguines nécessaires à la coagulation du sang). Elle est principalement héréditaire et rarement acquise (causée par une mutation génétique spontanée).
- Symptômes : tendance accrue aux saignements, ce qui peut facilement entraîner des saignements et des ecchymoses (hématomes). Le saignement prend également plus de temps que la normale. La gravité des symptômes dépend de la gravité de l'hémophilie.
- Diagnostic : Mesure de divers paramètres sanguins (aPTT, Quick value, temps de thrombine plasmatique, temps de saignement, nombre de plaquettes sanguines), détermination de l'activité des facteurs de coagulation
- Traitement : remplacement du facteur de coagulation manquant (sous forme de concentrés de facteurs) ; dans certains cas, d'autres médicaments (tels que la desmopressine pour l'hémophilie A légère)
Hémophilie : description
L'hémophilie est une maladie congénitale de la coagulation sanguine. Les personnes atteintes (hémophiles, « hémophiles ») ne peuvent pas former de facteurs de coagulation suffisamment fonctionnels. Ce sont des protéines dans le sang qui sont nécessaires à la coagulation du sang. En raison du manque de facteurs de coagulation, les caillots sanguins et les plaies ne peuvent pas se former facilement. Par conséquent, les patients hémophiles sont sujets aux saignements. Ils ont également des plaies qui saignent plus longtemps que la normale. Cela peut être dangereux dans certaines circonstances.
Le terme médical désignant un trouble de la coagulation sanguine avec une tendance accrue aux saignements est « diathèse hémorragique ». Le nom général d'un trouble de la coagulation est « coagulopathie ».
Hémophilie : fréquence
L'hémophilie survient presque exclusivement chez les garçons et les hommes. C'est relativement rare : seulement environ deux hommes sur 10 000 sont hémophiles. Il y a environ 10 000 personnes touchées en Allemagne. Environ 3 000 à 5 000 d'entre eux souffrent d'une forme sévère de la maladie.
Hémophilie : formes
Il existe différents facteurs de coagulation. Selon le facteur de coagulation affecté dans l'hémophilie, les professionnels de la santé font la distinction entre les différentes formes de la maladie.
L'hémophilie A.
Dans l'hémophilie A, il existe des problèmes avec le facteur de coagulation VIII (globuline antihémophilique A) : soit l'organisme ne peut pas le produire en quantité suffisante, soit il est défectueux. Environ 85 pour cent de tous les hémophiles souffrent d'hémophilie A. Presque tous sont des hommes.
L'hémophilie B.
Dans l'hémophilie B, le facteur de coagulation IX (globuline B antihémophilique ou facteur Christmas) est absent. Ici aussi, les patients sont majoritairement des hommes. Dans le passé, l'hémophilie B était plus fréquente dans la famille royale anglaise et dans la famille tsariste russe. C'est pourquoi on l'appelle aussi la « maladie des rois ».
Autres formes d'hémophilie
Outre les principales formes d'hémophilie A et B, il existe d'autres troubles héréditaires de la coagulation. L'un d'eux est le syndrome de von Willebrand-Jürgens, également appelé syndrome de von Willebrand (vWS). L'accent est mis ici sur le facteur de von Willebrand : ce facteur de coagulation est soit trop petit, soit défectueux. Comme pour l'hémophilie A et B, cela entraîne une tendance accrue aux saignements (diathèse hémorragique). En Allemagne, le syndrome de von Willebrand-Jürgens touche jusqu'à un pour cent de la population. Contrairement aux deux principales formes d'hémophilie, ce trouble de la coagulation touche autant les hommes que les femmes.
Dans de rares cas, d'autres troubles de la coagulation se produisent également, qui sont basés sur un manque de facteurs de coagulation. Ils ont également une tendance accrue à saigner (diathèse hémorragique). Ceux-ci inclus:
- Hémophilie C : Déficit en facteur XI fonctionnel
- Parahémophilie : absence de facteur V fonctionnel
- Hypoproconvertinémie : Déficit en facteur VII fonctionnel
- Déficit en facteur de Stuart Prower : manque de facteur de travail X
- Syndrome de Hageman : Déficit en facteur XII fonctionnel
- Déficit en fibrinase : déficit en facteur XIII fonctionnel
Hémophilie : symptômes
Les symptômes de l'hémophilie A (déficit en facteur VIII) et de l'hémophilie B (déficit en facteur IX) sont les mêmes - même si différents facteurs de coagulation sont affectés dans chaque cas. En général, on peut dire : moins il y a de facteurs de coagulation fonctionnels, plus le tableau clinique est prononcé.
Hémophilie : degrés de gravité
La gravité de l'hémophilie dépend de la réduction de l'activité des facteurs de coagulation par rapport à leur fonction chez les personnes en bonne santé. Si l'activité du facteur n'est que légèrement réduite, les personnes touchées ne présentent généralement aucun symptôme. En revanche, chez les hémophiles symptomatiques, l'activité du facteur est plus réduite, ce qui provoque des signes plus prononcés. Il existe trois degrés de gravité de l'hémophilie (A et B) :
Hémophilie légère:L'activité du facteur est de 6 à 45 pour cent de l'activité normale d'une personne en bonne santé ; environ 20 pour cent de toutes les personnes atteintes d'hémophilie ont cette gravité. Les personnes touchées ne remarquent souvent pas grand-chose de leur maladie. C'est pourquoi l'hémophilie n'est découverte chez de nombreuses personnes qu'à l'adolescence ou à l'âge adulte, lorsque les opérations ou les blessures graves saignent plus longtemps que prévu.
Hémophilie modérée : L'activité du facteur est de 1 à 5 pour cent de l'activité normale; environ 20 pour cent de tous les hémophiles sont également touchés. Les symptômes deviennent généralement évidents au cours des premières années de la vie avec des saignements inhabituellement longs et des ecchymoses fréquentes. Comme dans le cas de l'hémophilie légère, les saignements sont généralement le résultat de blessures ou d'une intervention chirurgicale. Les saignements spontanés sont rares.
Hémophilie sévère : L'activité du facteur est inférieure à 1 pour cent de l'activité normale. Cela s'applique à environ 55 pour cent de tous les hémophiles. L'hémophilie est généralement déjà perceptible à la naissance : l'ablation du cordon ombilical déclenche des saignements massifs. Les saignements de nez abondants ne sont pas rares chez les nourrissons. De plus, même les plus petites blessures ou bosses peuvent provoquer des saignements abondants sous la peau (ecchymoses). Les saignements internes sont également plus fréquents, par exemple des saignements douloureux dans les grosses articulations (comme les genoux, les coudes). En règle générale, de nombreux saignements n'ont pas de cause apparente (saignement spontané).
Hémophilie : risques et dangers
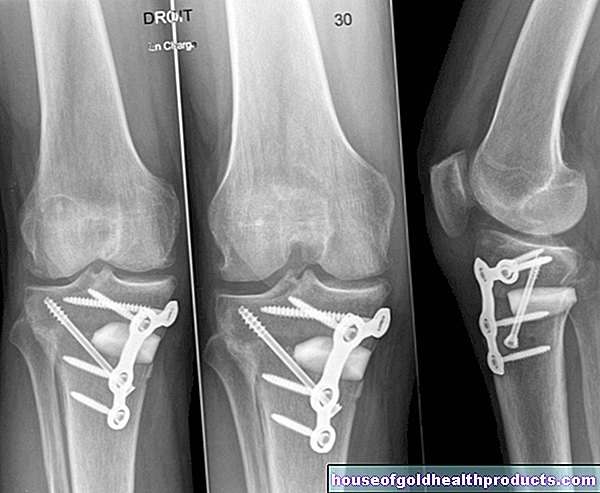
Les saignements dans les articulations (hémarthrose) se produisent souvent à plusieurs reprises, en particulier dans les cas d'hémophilie grave. En conséquence, l'articulation touchée peut se déformer, s'user prématurément (arthrose) et se raidir progressivement. Les personnes atteintes d'hémarthose avancée peuvent à peine bouger les bras et les jambes et dépendent d'un fauteuil roulant.
Un autre danger lié à l'hémophilie est le saignement dans les muscles : cela peut endommager le tissu musculaire et entraîner une faiblesse musculaire.
En principe, l'hémorragie interne dans l'hémophilie peut survenir dans n'importe quel organe. Les hémorragies cérébrales sont rares mais dangereuses : par exemple, elles peuvent altérer la capacité de réflexion et de concentration. Une hémorragie cérébrale sévère peut même être fatale ! Les saignements dans l'abdomen peuvent également mettre la vie en danger dans certaines circonstances. Des saignements abondants dans l'oropharynx peuvent affecter les voies respiratoires.
En plus des tendances hémorragiques (diathèse hémorragique), l'hémophilie, quel que soit son degré de gravité, peut également entraîner des troubles de la cicatrisation.
Syndrome de Von Willebrand-Juergens : Symptômes
Les personnes atteintes du syndrome de von Willebrand-Jürgens ont également une tendance accrue aux saignements. La plupart du temps, il y a des saignements légers sous la peau (ecchymoses), des saignements des gencives ou des saignements prolongés après une intervention chirurgicale, par exemple après une extraction dentaire. Mais il y a aussi des patients qui souffrent de saignements abondants.
Hémophilie : traitement
Le traitement de l'hémophilie dépend du type et de la gravité de l'hémophilie. Le médecin traitant établira un plan de traitement adapté à chaque patient. En plus du traitement médicamenteux, il peut également recommander des mesures générales. Par exemple, il peut être conseillé, notamment en cas d'hémophilie sévère, de prendre des précautions physiques et d'éviter certains sports (préjudiciables).
Hémophilie A et B
Aujourd'hui, des concentrés de facteur sont disponibles pour le traitement de l'hémophilie. Il s'agit de concentrés de facteurs de coagulation sanguine VIII (pour l'hémophilie A) ou IX (pour l'hémophilie B) obtenus à partir de plasma sanguin ou génétiquement modifiés. Ils doivent être injectés dans une veine (intraveineuse). De nombreux patients apprennent à s'injecter le concentré de facteur eux-mêmes. Cela leur donne beaucoup d'indépendance pour faire face à la maladie.
Dans les années 1960 et 1970, de nombreuses personnes en Allemagne ont contracté l'hépatite et/ou le VIH à cause de préparations de facteurs contaminés. Cela ne peut pratiquement plus se produire aujourd'hui : le plasma sanguin est strictement contrôlé et prétraité avant d'être administré. Et avec les concentrés de facteurs génétiquement modifiés, il n'y a généralement aucun risque d'infection.
En cas d'hémophilie légère et modérée, l'administration de concentré de facteur n'est nécessaire qu'en cas de nécessité (traitement à la demande) : L'anticoagulant est administré, par exemple, en cas de saignement plus abondant ou avant une opération envisagée. Les blessures mineures telles que les écorchures n'ont pas besoin d'être traitées avec un concentré de facteur. Le saignement peut généralement être arrêté en appliquant une légère pression sur la zone de saignement.
En revanche, les personnes atteintes d'hémophilie sévère doivent s'injecter régulièrement du concentré de facteur (traitement de longue durée). Le facteur VIII dans l'hémophilie A est administré deux à trois fois par semaine. Le facteur IX dans l'hémophilie B n'a généralement besoin d'être injecté qu'une à deux fois par semaine en raison de son temps de rétention plus long dans le sang.
Chirurgies et blessures aiguës
Avant les opérations (même si une extraction dentaire est prévue), un traitement préparatoire doit être réalisé pour tous les patients hémophiles. Un concentré de facteur est généralement administré à cette fin. C'est le seul moyen de prévenir les complications graves dues à une perte de sang excessive pendant la chirurgie.
Dans le cas de l'hémophilie A légère, au lieu de concentrés de facteur, un médicament qui stabilise la coagulation sanguine peut être administré avant une opération planifiée. Cela inclut la desmopressine, par exemple. Il s'agit d'une protéine produite artificiellement. Il stimule la libération du facteur VIII stocké par les vaisseaux sanguins. Le médicament ne peut être utilisé que pendant quelques jours, sinon les magasins seront bientôt vides.
Conseil : Avant une intervention planifiée, les personnes atteintes d'hémophilie doivent discuter avec leur hématologue ou centre d'hémophilie du traitement préventif à conseiller dans leur cas.
En cas de blessures aiguës en urgence, en plus des mesures locales d'hémostase (par exemple des bandages compressifs), l'administration de concentré de facteur est également nécessaire.
Concentré de facteur : complications
Certaines personnes forment des anticorps (inhibiteurs) contre les facteurs de coagulation contenus dans le concentré de facteur. Cette hémophilie dite inhibiteur est significativement plus fréquente chez les personnes atteintes d'hémophilie A que chez les personnes atteintes d'hémophilie B. Les inhibiteurs inactivent le facteur de coagulation ajouté. La thérapie n'est alors pas aussi efficace que souhaité. L'hémophilie B menace également de graves réactions allergiques et d'autres complications.
La quantité d'inhibiteurs dans le sang est indiquée dans l'unité dite Bethesda (BE). Plus la valeur BE est élevée, plus il y a d'inhibiteurs dans le sang du patient.
Dans l'hémophilie A, une légère accumulation d'inhibiteurs peut souvent être compensée en augmentant la dose de concentré de facteur. Si des inhibiteurs sont formés en grande quantité, un traitement de tolérance immunitaire est recommandé : le patient reçoit des doses très élevées du facteur de coagulation manquant dans un schéma thérapeutique complexe. Le système immunitaire doit progressivement s'habituer à sa présence et arrêter la formation d'inhibiteurs.
L'hémophilie inhibiteur rare de l'hémophilie B est traitée différemment. Par exemple, les personnes touchées reçoivent des médicaments qui affectent le système immunitaire (immunomodulateurs).
Anti douleur
L'hémophilie grave peut causer une douleur intense aux personnes touchées. Par exemple, les saignements dans les articulations peuvent être très douloureux. Ensuite, les analgésiques comme l'ibuprofène aident. En revanche, l'analgésique acide acétylsalicylique (AAS) n'est pas adapté à l'hémophilie : il augmente encore la tendance aux saignements (effet secondaire de l'AAS).
Syndrome de von Willebrand-Juergens (VWS)
Dans le syndrome de von Willebrand-Jürgens, une distinction est faite entre différents types, qui sont traités différemment : Dans ce qu'on appelle le type 1, le principe actif desmopressine est administré lorsque cela est nécessaire (avant une opération ou en cas de saignement aigu). Il stimule la libération des facteurs de coagulation stockés.
La desmopressine est également utilisée dans le type 2; cependant, le médicament ne fonctionne pas toujours ici. Ensuite, le médecin prescrit un concentré de facteur (avec facteur de von Willebrand) à la place.
Les patients atteints de VWS de type 3 sont toujours traités avec un concentré de facteur.
Hémophilie : causes et facteurs de risque
L'hémophilie est une maladie génétique congénitale qui est généralement héréditaire. Elle survient plus rarement spontanément (à la suite d'un changement génique spontané = mutation spontanée).
Chez les hémophiles, l'information génétique requise pour produire un facteur de coagulation fonctionnel est erronée : dans l'hémophilie A, il s'agit du facteur VIII de coagulation, dans l'hémophilie B, du facteur IX. Le résultat d'un mauvais plan de construction est que le facteur de coagulation pertinent ne peut pas être produit en une quantité suffisamment fonctionnelle. Cela perturbe la coagulation du sang : les plaies ne se referment pas aussi rapidement, le saignement prend donc un temps inhabituellement long. Certains patients présentent également des saignements spontanés (sans cause apparente).
Hémophilie A et B : héréditaire
Les plans (gènes) de toutes les parties du corps se trouvent sur les chromosomes. Dans le noyau de chaque cellule du corps, il y a 46 chromosomes, dont deux chromosomes sexuels, qui, entre autres, déterminent le sexe. Les femmes ont deux chromosomes sexuels X (XX) : un chromosome X chacun a été hérité de la mère et un du père. Les hommes, en revanche, ont un chromosome Y hérité du père et un chromosome X hérité de la mère (XY).
Les gènes des facteurs de coagulation se trouvent sur le chromosome X. Chez les femmes qui ont deux chromosomes X, si l'un d'eux contient un modèle défectueux pour un facteur de coagulation, cela peut généralement être compensé par l'autre chromosome X. Ils sont donc en grande partie asymptomatiques tout au long de leur vie.
Si une telle femme a un enfant, elle transmet l'un des deux chromosomes X à la progéniture. La probabilité qu'il s'agisse de la copie avec le plan défectueux est de 50 %. En transmettant le défaut génétique, la mère devient la porteuse (porteuse) de l'hémophilie. S'il y a un fils, il naîtra hémophile. Une fille, en revanche, devient généralement une porteuse potentielle à son tour.
Dans de rares cas, le facteur de coagulation pertinent n'est pas suffisamment formé, même chez les conducteurs féminins. Les blessures causées par des blessures ou des opérations peuvent alors saigner pendant longtemps. Il est encore plus rare qu'une fille hérite d'un chromosome X défectueux des deux parents. Ceci est principalement en relation avec la maladie héréditaire du syndrome de Turner. Les filles touchées présentent le tableau complet de l'hémophilie.
Syndrome de von Willebrand-Juergens
Dans le syndrome de von Willebrand-Jürgens, les instructions pour le facteur de von Willebrand (vWF) montrent une mutation : le facteur de coagulation est trop petit ou est défectueux. La mutation du gène peut survenir aussi bien chez les hommes que chez les femmes.
Hémophilie : examens et diagnostic
Si quelqu'un a des saignements spontanés fréquents (comme des saignements de nez) ou des ecchymoses très facilement, cela pourrait être une indication d'hémophilie. Ce soupçon est particulièrement évident si la famille a déjà connu des cas d'hémophilie. Les personnes touchées devraient avoir une tendance accrue aux saignements clarifiée par un médecin. Le premier point de contact si vous suspectez une hémophilie est votre médecin de famille :
Le médecin prendra d'abord les antécédents médicaux du patient (anamnèse) en discutant avec le patient : il présente les symptômes décrits en détail, s'enquiert des maladies sous-jacentes et s'il existe des cas connus d'hémophilie dans la famille.
Les tests de laboratoire sont particulièrement importants pour élucider une hémophilie possible. Le médecin prélève un échantillon de sang sur le patient afin de le faire examiner en laboratoire pour divers paramètres : Dans l'hémophilie, le temps dit aPTT (temps de céphaline activée) est plus long que chez les personnes en bonne santé. En revanche, la valeur dite de Quick (temps de thromboplastine, TPZ) et le temps de thrombine plasmatique (PTZ) sont généralement normaux ; elles ne se prolongent que dans l'hémophilie sévère. Le nombre de plaquettes sanguines (thrombocytes) et le temps dit de saignement sont également normaux dans les hémophilies A et B. Dans le syndrome de von Willebrand-Jürgens, en revanche, le temps de saignement est prolongé. Le temps de saignement est le temps qu'il faut pour que le saignement s'arrête.
Afin de pouvoir déterminer sans équivoque l'hémophilie A ou B, l'activité des facteurs de coagulation (VIII, IX) doit être analysée. Un spécialiste en hématologie ou un centre médical spécialisé (centre d'hémophilie) en est responsable.
Hémophilie : test chez les nouveau-nés et les bébés à naître
Si une famille a déjà développé une hémophilie, la coagulation des nouveau-nés de sexe masculin est généralement contrôlée immédiatement après la naissance. De cette façon, l'hémophilie peut être détectée à un stade précoce. Vous pouvez également rechercher une hémophilie pendant la grossesse.
Si une femme soupçonne qu'elle a une prédisposition génétique à l'hémophilie et qu'elle est donc une porteuse potentielle, cela peut être clarifié par un test génétique.
Hémophilie : évolution de la maladie et pronostic
L'hémophilie n'est pas encore guérie. Les patients doivent composer avec le manque de facteurs de coagulation tout au long de leur vie. Avec l'aide de concentrés de facteur, cependant, ils peuvent généralement mener une vie en grande partie normale.
Si elle n'est pas traitée, l'hémophilie modérée et sévère entraîne souvent de graves complications. Par exemple, des saignements dans les muscles peuvent causer des dommages musculaires. Une hémorragie dans les articulations peut entraîner de l'arthrose et un raidissement des articulations. De telles complications peuvent être évitées si l'hémophilie est détectée tôt et traitée de manière cohérente.
Mots Clés: dormir systèmes d'organes tcm










.jpg)