Le syndrome de Marfan
Mareike Müller est rédactrice indépendante au service médical et médecin assistante en neurochirurgie à Düsseldorf. Elle a étudié la médecine humaine à Magdebourg et a acquis une grande expérience médicale pratique lors de ses séjours à l'étranger sur quatre continents différents.
En savoir plus sur les experts Tout le contenu de est vérifié par des journalistes médicaux.Le syndrome de Marfan (SFM) est une maladie génétique du tissu conjonctif. Les patients présentent des symptômes différents à des degrés divers : doigts longs et membres longs et étroits ou lésions des vaisseaux sanguins. Il n'y a pas de remède pour le syndrome de Marfan. Des contrôles réguliers peuvent prévenir les complications. Lisez tout sur le syndrome de Marfan ici !
Codes CIM pour cette maladie : Les codes CIM sont des codes internationalement reconnus pour les diagnostics médicaux. On les retrouve, par exemple, dans les lettres des médecins ou sur les certificats d'incapacité de travail. Q87
Syndrome de Marfan : description
Le syndrome de Marfan est une maladie génétique qui se transmet des parents à l'enfant ou se développe spontanément. Une maladie qui se développe spontanément est également connue sous le nom de maladie sporadique. Cela s'applique à environ 25 à 30 pour cent des patients atteints du syndrome de Marfan. Dans l'ensemble, une à cinq personnes sur 10 000 dans la population sont touchées par le syndrome de Marfan. Il n'y a pas de différence entre les sexes.
Syndrome de Marfan : Symptômes
Les signes du syndrome de Marfan sont très différents et différemment prononcés selon les patients. Même dans la même famille, les symptômes du syndrome de Marfan peuvent différer considérablement parmi les membres de la famille malades. Divers systèmes organiques sont touchés par la maladie. Les plus courants sont les changements
- Système cardiovasculaire
- squelette
- œil
Syndrome de Marfan : Système cardiovasculaire
Les patients atteints du syndrome de Marfan présentent un risque accru de mort subite. La raison en est une déchirure fréquente de la paroi de l'artère principale (dissection aortique). En raison de la formation d'un espace dans la paroi aortique, le sang n'est plus transporté dans les petits vaisseaux sanguins, mais s'infiltre plutôt dans les espaces. Le risque de dissection aortique est accru chez les patients atteints du syndrome de Marfan car leur aorte, dont les parois sont fragilisées, s'élargit progressivement (dilatation aortique progressive).
De plus, les patients souffrent souvent de lésions des valves cardiaques telles qu'une régurgitation aortique et mitrale. Ceux-ci peuvent entraîner des arythmies cardiaques. De plus, ils sont à risque d'inflammation du cœur (endocardite) et d'insuffisance cardiaque.
Syndrome de Marfan : Squelette
Les changements squelettiques sont souvent le premier signe du syndrome de Marfan. Les patients se distinguent par leur grande taille et leurs extrémités très étroites et longues. Le doigté d'araignée (arachnodactylie) est un symptôme bien connu. Les doigts d'araignée sont appelés ainsi parce qu'ils sont extrêmement longs et étroits.
En outre, de nombreux patients présentent des malformations thoraciques telles qu'une poitrine de poulet ou en entonnoir. En tant que modifications squelettiques supplémentaires, ils souffrent souvent de scoliose, d'une flexion et d'une torsion de la colonne vertébrale. De plus, certains patients ont des os du visage sous-développés, comme les pommettes ou la mâchoire supérieure.
L'ensemble de ces changements squelettiques est également connu sous le nom d'habitus marfanoïde.
Syndrome de Marfan : œil
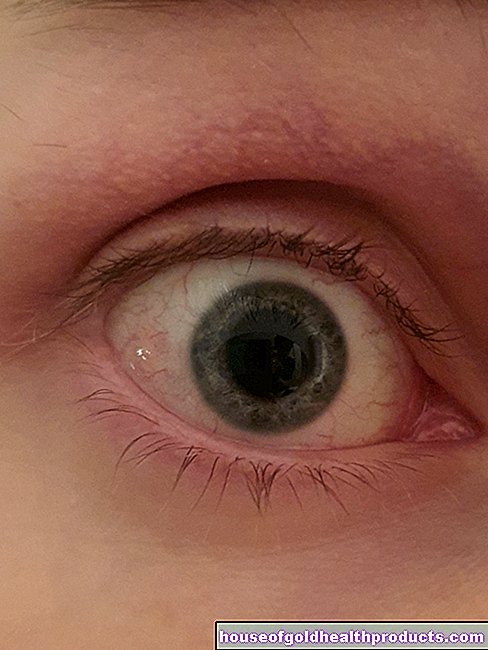
Les modifications de l'œil causées par le syndrome de Marfan affectent principalement le cristallin. Elle est souvent décalée (ectopie du cristallin). Cela menace le patient de cécité. Un autre facteur de risque de cécité est la myopie. Elle est causée par un globe oculaire trop long. Ce changement peut également conduire à un décollement de la rétine.
Syndrome de Marfan : symptômes affectant d'autres organes
En plus des systèmes organiques mentionnés, le syndrome de Marfan peut également endommager d'autres structures. Cela inclut les poumons, entre autres. Les personnes touchées ont un risque accru de développer un pneumothorax. Les médecins comprennent que cela signifie le détachement de la plèvre pulmonaire de la plèvre et la pénétration d'air dans cet espace. Cette condition peut être mortelle car les poumons s'effondrent dans la zone touchée.
Les vergetures sont souvent observées sur la peau des patients atteints du syndrome de Marfan comme signe de faiblesse du tissu conjonctif.
Au cours de la vie, une soi-disant duraectasie peut se développer. Il s'agit d'une extension des méninges, généralement au niveau de la colonne lombaire. Il est souvent asymptomatique. Dans certains cas, il peut causer de la douleur lorsque le sac dural appuie sur les nerfs spinaux sortants.
Syndrome de Marfan : causes et facteurs de risque
Le syndrome de Marfan est une maladie héréditaire autosomique dominante. Cela signifie qu'il y a un changement (mutation) dans un gène de notre constitution génétique qui déclenche la maladie. Autosomique dominant décrit que cette information génétique est située sur un complexe génique non spécifique au genre (autosomique) et apparaît toujours (dominante).
Lorsqu'un patient atteint du syndrome de Marfan a un enfant, il peut hériter du gène malade ou sain. Parce que chaque personne a une double constitution génétique. Cela signifie que la probabilité de transmission est de 50 pour cent. Un enfant d'un patient atteint du syndrome de Marfan a 50% de chances de contracter la maladie.
Syndrome de Marfan : tissu conjonctif endommagé
La mutation à l'origine du syndrome de Marfan se situe sur le bras long du chromosome 15 (15q21). Il affecte le soi-disant gène FBN1. Ce gène est responsable de la formation d'une protéine du tissu conjonctif, la fibrilline-1. La fibrilline-1 est importante pour la stabilité du tissu conjonctif. Si sa formation est restreinte par la mutation, le tissu conjonctif perd sa stabilité.
Syndrome de Marfan : diverses formes
La gravité du syndrome de Marfan varie. Les médecins parlent alors d'expressivité variable. Cela signifie que les symptômes des patients sont également différents au sein d'une famille. Malgré la même mutation, un patient peut à peine avoir des symptômes, tandis qu'un frère montre l'image complète du syndrome de Marfan.
Syndrome de Marfan : Investigations et diagnostic
Le diagnostic du syndrome de Marfan est souvent posé par un pédiatre. Dans l'ensemble, divers spécialistes jouent un rôle dans le diagnostic, le traitement et le conseil. En plus du pédiatre, cela comprend les généticiens humains, les cardiologues, les orthopédistes et les ophtalmologistes. Avant que le diagnostic ne soit définitivement posé, votre médecin vous demandera d'abord en détail vos antécédents médicaux (anamnèse). Il vous posera entre autres les questions possibles suivantes :
- Un membre de la famille a-t-il le syndrome de Marfan ?
- Sentez-vous parfois un cœur qui s'emballe?
- Avez-vous toujours été plus grand que les autres quand vous étiez enfant ?
- Êtes-vous myope ?
Examen physique du syndrome de Marfan
Votre médecin procédera alors à un examen physique. Ce faisant, il examine d'abord de près le squelette. Il fait attention à la longueur des os individuels, à la forme de la poitrine et à la forme du visage. Puis il écoute le cœur et les poumons. Des arythmies cardiaques ou des bruits d'écoulement peuvent être remarqués au-dessus de l'artère principale.
Afin de faire le diagnostic du syndrome de Marfan, les critères dits de Gent ont été développés. Il répertorie divers symptômes de la maladie sous différentes formes. Lorsqu'un certain nombre de critères sont réunis, le diagnostic peut être posé.
Un test génétique du syndrome de Marfan est également possible. La constitution génétique est analysée et la mutation responsable de la maladie est recherchée. S'il existe des cas de syndrome de Marfan dans la famille, un diagnostic approprié peut être posé avant la naissance.
Syndrome de Marfan : Images cliniques similaires
D'autres maladies génétiques pouvant entraîner des symptômes similaires doivent être distinguées du syndrome de Marfan. Il s'agit, entre autres,
- Syndrome d'Ehlers-Danlos
- Syndrome de Loeys-Dietz
- Syndrome de Sphintzen-Goldberg
- Syndrome de MASSE
Syndrome de Marfan : Traitement
Le syndrome de Marfan étant une maladie génétique, la cause elle-même, la mutation, ne peut être traitée. Le but de la thérapie est un contrôle régulier du patient par divers spécialistes afin de prévenir les complications. Plus important encore, la surveillance cardiaque est importante pour prévenir la mort subite par dissection aortique. Ceci peut être réalisé en contrecarrant la dilatation aortique en administrant des bêta-bloquants et en limitant l'activité physique. La dilatation aortique peut être contrôlée par des examens échographiques annuels et la racine aortique peut être corrigée à temps avant la dissection.
Autres opérations qui peuvent être nécessaires dans le cas du syndrome de Marfan
- Correction de la scoliose
- Correction de la poitrine
- Retrait de lentille
Syndrome de Marfan: évolution de la maladie et pronostic
La probabilité que la mutation soit transmise d'un parent à un enfant est de 50 %. Les couples avec un partenaire atteint du syndrome de Marfan qui envisagent d'avoir des enfants devraient demander conseil à un généticien humain.
Aujourd'hui, l'espérance de vie et la qualité de vie sont presque illimitées pour les patients atteints du syndrome de Marfan. Alors que l'espérance de vie était clairement limitée dans le passé, elle a augmenté de 30 ans au cours des 30 dernières années. Cependant, les patients présentent toujours un risque accru de dissection aortique, ce qui peut entraîner une mort subite. La dissection aortique est le plus souvent observée vers l'âge de 30 ans. Des contrôles réguliers par les spécialistes traitants peuvent réduire le risque de dissection aortique dans le syndrome de Marfan.
Mots Clés: diète médecine palliative remèdes maison